Data Processing
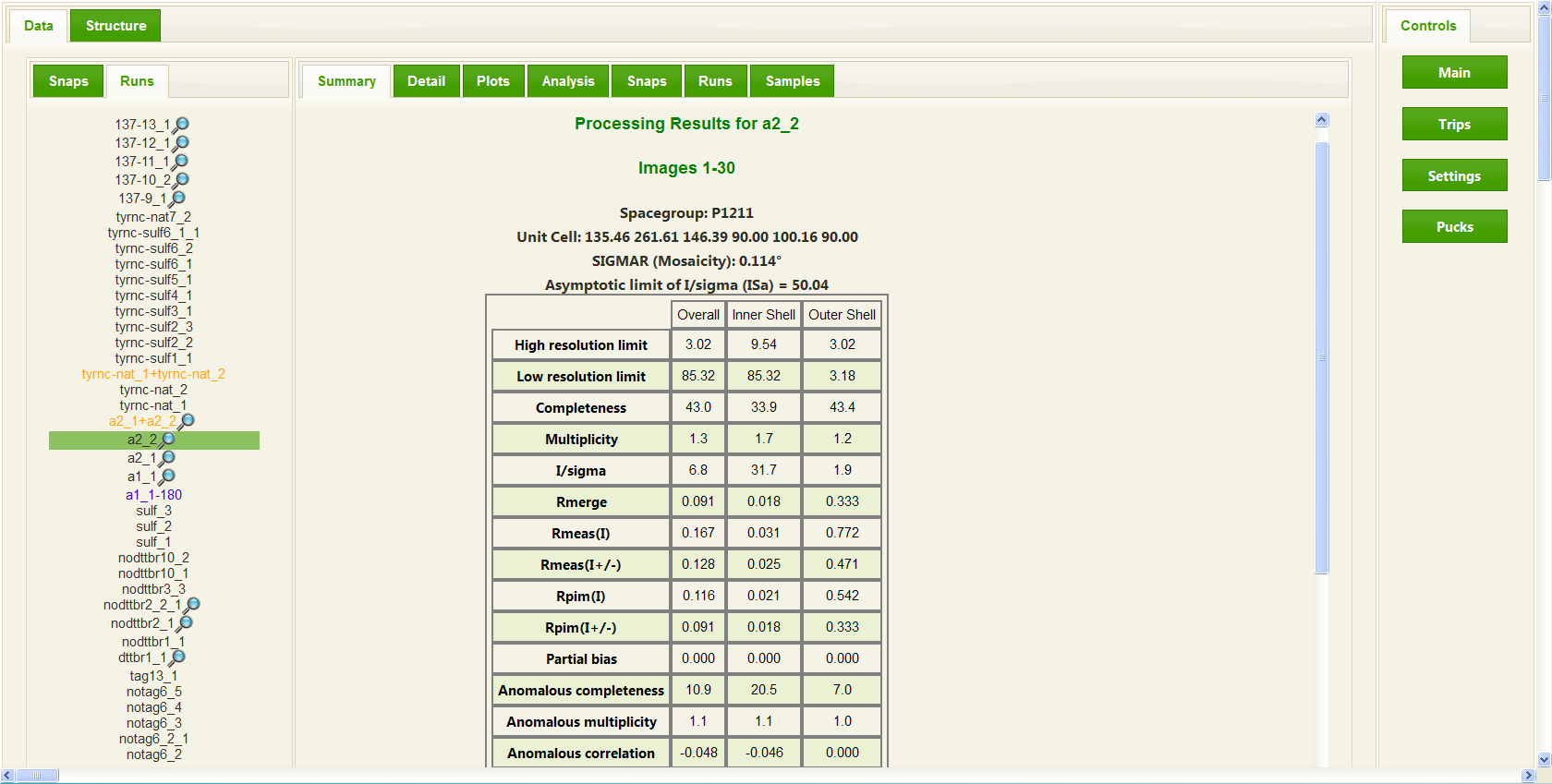
In the case of a collected data set, RAPD will automatically initiate integration and scaling of the data set. In most cases this data processing begins while the data is being collected, with results displayed and updated throughout the data collection. RAPD utilizes the program XDS to both integrate and scale the data and then uses the CCP4 programs Pointless and Scala to provide a summary of the results in a format most users are familiar with. On the user interface, a summary for a data collection run consists of a table providing common measures of data quality such as I/sigma, Rmerge, Rmeas, and Rpim in a table format showing these values for the overall data set, the inner resolution shell, and the outer resolution shell. In addition the summary provides the user with some feedback on whether an anomalous signal is present in the data collected by presenting the anomalous correlation and anomalous slope. More detailed information is available to the users on additional tabs, including plots of the integration and scaling results. Once a data set is completed, further analysis is performed by comparing the unit cell parameters to entries already deposited in the Protein Data Bank (PDB), running the phenix.xtriage reflection analysis utility (and presenting those results), and generating a self-rotation patterson map to aid in idenitfying non-crystallographic symmetries.